Proteoform inference: reproducing Bludau et al. (2021)
This notebook shows how to use ProteoPy for proteoform group inference following the approach of Bludau et al. (2021):
Bludau I, Frank M, Dörig C, Cai Y, Heusel M, Rosenberger G, Picotti P, Collins BC, Röst H, and Aebersold R. Systematic detection of functional proteoform groups from bottom-up proteomic datasets. Nature Communications, 12:3810, 2021. doi:10.1038/s41467-021-24030-x
The authors introduced COPF (COrrelation-based functional ProteoForm assessment), a data-driven strategy that assigns peptides to co-varying proteoform groups based on peptide correlation patterns. They applied it to SWATH-MS (DIA) data from five mouse tissues (brain, brown adipose tissue, heart, liver, and quadriceps) across eight BXD mice, originally published by Williams et al. (2018):
Williams EG, Wu Y, Wolski W, Kim JY, Lan J, Hasan M, Halter C, Jha P, Ryu D, Auwerx J, and Aebersold R. Quantifying and Localizing the Mitochondrial Proteome Across Five Tissues in A Mouse Population. Molecular & Cellular Proteomics, 17(9):1766–1777, 2018. doi:10.1074/mcp.RA118.000554
Here, we show that the COPF implementation in ProteoPy can reproduce the core proteoform inference workflow and selected results (Figures 6B and 7A/C/D/F).
Setup
[1]:
# Jupyter autoreload for development
%load_ext autoreload
%autoreload 2
[2]:
import os
import random
from pathlib import Path
import numpy as np
import pandas as pd
import anndata as ad
import scanpy as sc
import matplotlib as mpl
import matplotlib.pyplot as plt
from matplotlib.pyplot import rc_context
import proteopy as pr # Convention: import proteopy as pr
from proteopy.utils import is_proteodata
random.seed(42)
cwd = Path(".").resolve()
root = cwd.parents[1]
os.chdir(root)
/home/ifichtner/miniforge3/envs/proteopy-usage2/lib/python3.10/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
Reading in the data
The Williams et al. (2018) dataset contains peptide-level SWATH-MS intensities from five tissues of eight BXD mice. ProteoPy provides a built-in download function (pr.download.williams_2018()) that fetches the dataset from the original supplementary archive, processes it, and saves it as three separate files:
Intensities — long-format table with columns
sample_id,peptide_id, andintensity(one row per measurement)Sample annotation — maps each
sample_idto itstissueandmouse_idPeptide annotation — maps each
peptide_idto itsprotein_idandgene_id
This separation into three files mirrors a common data delivery format in proteomics. The files are then read into an AnnData object using pr.read.long().
[3]:
intensities_path = "data/williams-2018_mouse-tissue_intensities.tsv"
sample_annotation_path = "data/williams-2018_mouse-tissue_sample_annotation.tsv"
peptide_annotation_path = "data/williams-2018_mouse-tissue_peptide_annotation.tsv"
pr.download.williams_2018(
intensities_path=intensities_path,
sample_annotation_path=sample_annotation_path,
var_annotation_path=peptide_annotation_path,
force=True, # Overwrite files if already exist
)
Downloading data from 'https://ars.els-cdn.com/content/image/1-s2.0-S1535947620320569-mmc1.zip' to file '/home/ifichtner/.cache/proteopy/williams_2018_mmc1.zip'.
A quick look at the first rows of each file confirms the expected structure:
[4]:
!head -n5 {intensities_path}
sample_id peptide_id intensity
C57_Brain AAAAAAAAAAAAAAAGAAGK 26302.0
DBA_Brain AAAAAAAAAAAAAAAGAAGK 15460.0
45_Brain AAAAAAAAAAAAAAAGAAGK 24631.0
66_Brain AAAAAAAAAAAAAAAGAAGK 26554.0
[5]:
!head -n5 {sample_annotation_path}
sample_id tissue mouse_id
C57_Brain Brain C57
DBA_Brain Brain DBA
45_Brain Brain 45
66_Brain Brain 66
[6]:
!head -n5 {peptide_annotation_path}
peptide_id protein_id gene_id
AAAAAAAAAAAAAAAGAAGK P55012 Slc12a2
AAAAADLANR Q9JHS4 Clpx
AAAADGEPLHNEEER Q80WW9 Ddrgk1
AAAAEGARPLER Q80UM7 Mogs
With the three files on disk, pr.read.long() reads the long-format intensities, pivots them into a dense matrix, and merges the sample and peptide annotations into a single AnnData object. Setting fill_na=0 replaces any missing intensity values with zero.
[7]:
adata = pr.read.long(
intensities=intensities_path,
sample_annotation=sample_annotation_path,
var_annotation=peptide_annotation_path,
level="peptide",
fill_na=0,
)
adata
[7]:
AnnData object with n_obs × n_vars = 40 × 32690
obs: 'sample_id', 'tissue', 'mouse_id'
var: 'peptide_id', 'protein_id', 'protein_id_annotation', 'gene_id'
[8]:
print("Is proteodata: ", is_proteodata(adata))
Is proteodata: (True, 'peptide')
[9]:
adata.obs.head(n=10)
[9]:
| sample_id | tissue | mouse_id | |
|---|---|---|---|
| 101_Brain | 101_Brain | Brain | 101 |
| 45_Brain | 45_Brain | Brain | 45 |
| 66_Brain | 66_Brain | Brain | 66 |
| 68_Brain | 68_Brain | Brain | 68 |
| 73_Brain | 73_Brain | Brain | 73 |
| 80_Brain | 80_Brain | Brain | 80 |
| BAT_101 | BAT_101 | BAT | 101 |
| BAT_45 | BAT_45 | BAT | 45 |
| BAT_66 | BAT_66 | BAT | 66 |
| BAT_68 | BAT_68 | BAT | 68 |
[10]:
adata.var.head(n=10)
[10]:
| peptide_id | protein_id | protein_id_annotation | gene_id | |
|---|---|---|---|---|
| AAAAAAAAAAAAAAAGAAGK | AAAAAAAAAAAAAAAGAAGK | P55012 | P55012 | Slc12a2 |
| AAAAADLANR | AAAAADLANR | Q9JHS4 | Q9JHS4 | Clpx |
| AAAADGEPLHNEEER | AAAADGEPLHNEEER | Q80WW9 | Q80WW9 | Ddrgk1 |
| AAAAEGARPLER | AAAAEGARPLER | Q80UM7 | Q80UM7 | Mogs |
| AAAAKEEAPK | AAAAKEEAPK | Q91XV3 | Q91XV3 | Basp1 |
| AAAANLC(UniMod:4)PGDVILAIDGFGTESMTHADAQDR | AAAANLC(UniMod:4)PGDVILAIDGFGTESMTHADAQDR | O70209 | O70209 | Pdlim3 |
| AAAAYALGR | AAAAYALGR | Q6ZQ73 | Q6ZQ73 | Cand2 |
| AAADLM(UniMod:35)AYC(UniMod:4)EAHAKEDPLLTPVPASENPFR | AAADLM(UniMod:35)AYC(UniMod:4)EAHAKEDPLLTPVPAS... | P63213 | P63213 | Gng2 |
| AAADLMAYC(UniMod:4)EAHAK | AAADLMAYC(UniMod:4)EAHAK | P63213 | P63213 | Gng2 |
| AAADLMAYC(UniMod:4)EAHAKEDPLLTPVPASENPFR | AAADLMAYC(UniMod:4)EAHAKEDPLLTPVPASENPFR | P63213 | P63213 | Gng2 |
Define consistent color schemes for sample annotation metadata
[11]:
# Tissues
n_tissues = adata.obs["tissue"].nunique()
cmap = mpl.colormaps["Set2"]
adata.uns["colors_tissue"] = cmap(range(n_tissues)).tolist()
tissue_order = ["Brain", "BAT", "Heart", "Liver", "Quad"]
# BXD mice
n_mice = adata.obs["mouse_id"].nunique()
cmap = mpl.colormaps["tab10"]
adata.uns["colors_mouse_id"] = cmap(range(n_mice)).tolist()
Quality control and preprocessing
By study design, the dataset includes 5 tissues derived from 8 BXD mice:
[12]:
pr.pl.n_samples_per_category(
adata,
category_key="tissue",
order=tissue_order,
color_scheme=adata.uns["colors_tissue"],
)
[13]:
pr.pl.n_samples_per_category(
adata,
category_key="mouse_id",
color_scheme=adata.uns["colors_mouse_id"],
)
Similar to the original analysis, we remove peptides from the iRT protein, which was used for calibration, and A2ASS6, which has extraordinarily many peptides and distorts the analysis.
[14]:
irt_mask = (adata.var["protein_id"] == "iRT_protein")
adata = adata[:, ~irt_mask]
[15]:
A2ASS6_mask = (adata.var["protein_id"] == "A2ASS6")
print(f"N peptides for protein A2ASS6: {A2ASS6_mask.sum()}")
N peptides for protein A2ASS6: 919
[16]:
adata = adata[:, ~A2ASS6_mask]
Next, peptides that differ only by technical modifications or derived from missed cleavages are summarized by summing up their intensities.
[17]:
pr.pp.summarize_modifications(adata, method="sum", verbose=True)
pr.pp.summarize_overlapping_peptides(adata)
Stripping modifications: 31760 peptides -> 29862 unique stripped sequences (method='sum').
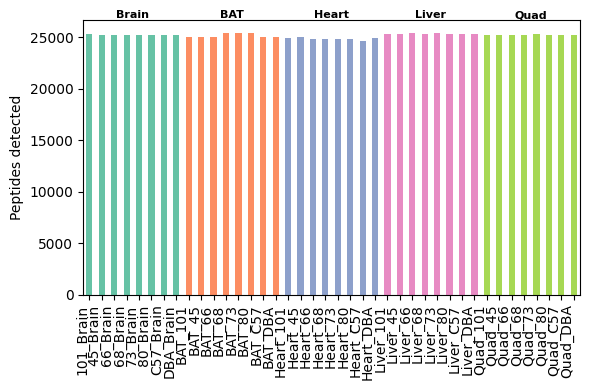
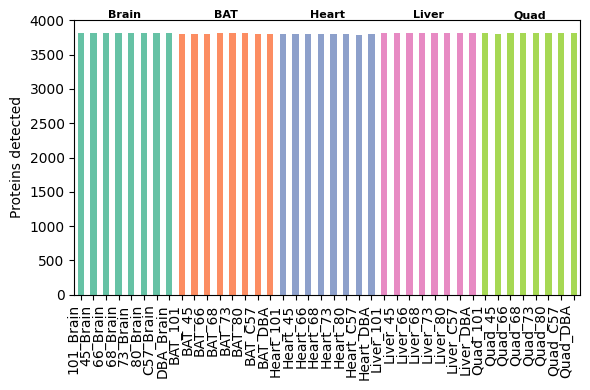
After these preprocessing steps, the dataset includes approximately 25,000 peptides and 3,800 proteins per sample.
[18]:
pr.pl.n_peptides_per_sample(
adata,
zero_to_na=True,
order_by="tissue",
order=tissue_order,
color_scheme=adata.uns["colors_tissue"],
print_stats=True,
)
Global:
mean_count std_count median_count min_count max_count mean_pct std_pct median_pct min_pct max_pct
25184.2 199.1 25258.5 24623 25418 98.8 0.8 99.1 96.6 99.7
Per tissue:
tissue mean_count std_count median_count min_count max_count mean_pct std_pct median_pct min_pct max_pct
BAT 25181.2 191.5 25077.5 24997 25418 98.8 0.8 98.4 98.0 99.7
Brain 25269.4 7.2 25268.5 25261 25280 99.1 0.0 99.1 99.1 99.1
Heart 24860.5 110.9 24858.0 24623 24996 97.5 0.4 97.5 96.6 98.0
Liver 25369.6 11.8 25372.0 25352 25386 99.5 0.0 99.5 99.4 99.6
Quad 25240.0 27.7 25240.0 25211 25289 99.0 0.1 99.0 98.9 99.2
[19]:
pr.pl.n_proteins_per_sample(
adata,
zero_to_na=True,
order_by="tissue",
order=tissue_order,
color_scheme=adata.uns["colors_tissue"],
print_stats=True,
)
Global:
mean_count std_count median_count min_count max_count mean_pct std_pct median_pct min_pct max_pct
3808.8 5.8 3811.5 3790 3815 99.8 0.2 99.9 99.3 99.9
Per tissue:
tissue mean_count std_count median_count min_count max_count mean_pct std_pct median_pct min_pct max_pct
BAT 3807.1 5.3 3805.0 3801 3814 99.7 0.1 99.7 99.6 99.9
Brain 3812.1 0.6 3812.0 3811 3813 99.9 0.0 99.9 99.8 99.9
Heart 3800.1 4.6 3801.0 3790 3806 99.6 0.1 99.6 99.3 99.7
Liver 3814.0 0.5 3814.0 3813 3815 99.9 0.0 99.9 99.9 99.9
Quad 3810.8 1.5 3811.0 3808 3813 99.8 0.0 99.8 99.8 99.9
[20]:
pr.pl.n_peptides_per_protein(adata, print_stats=True)
mean median mode variance min max
6.680115 4.0 1 70.811955 1 118
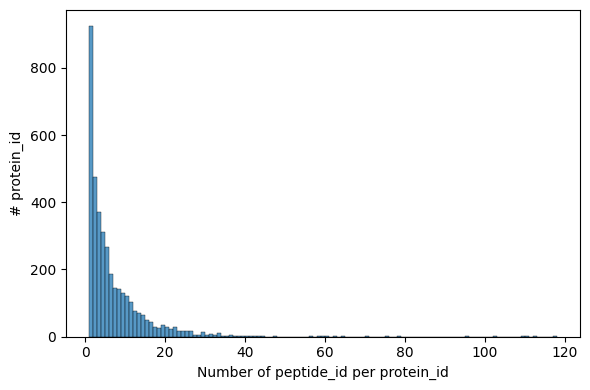
[20]:
<Axes: xlabel='Number of peptide_id per protein_id', ylabel='# protein_id'>
Note that no log transformation, data normalization or missing value imputation is performed here. This is because of the overall very high data consistency across the dataset and because COPF is based on covariation rather than direct intensity differences across conditions. This is also in agreement with the analysis by Bludau et al. (2021). However, other datasets might require additional preprocessing steps.
Exploratory analysis of peptide-level data
Before proteoform inference, ProteoPy can be used to perform classical exploratory data analysis steps, for example to investigate the primary sources of variation in the dataset.
Sample correlation matrix
High intra-tissue correlation and lower inter-tissue correlation show that peptide intensities are strongly driven by the tissue of origin and not by the mouse strain.
[21]:
pr.pl.sample_correlation_matrix(
adata,
method="pearson",
margin_color="tissue",
color_scheme=adata.uns["colors_tissue"],
)
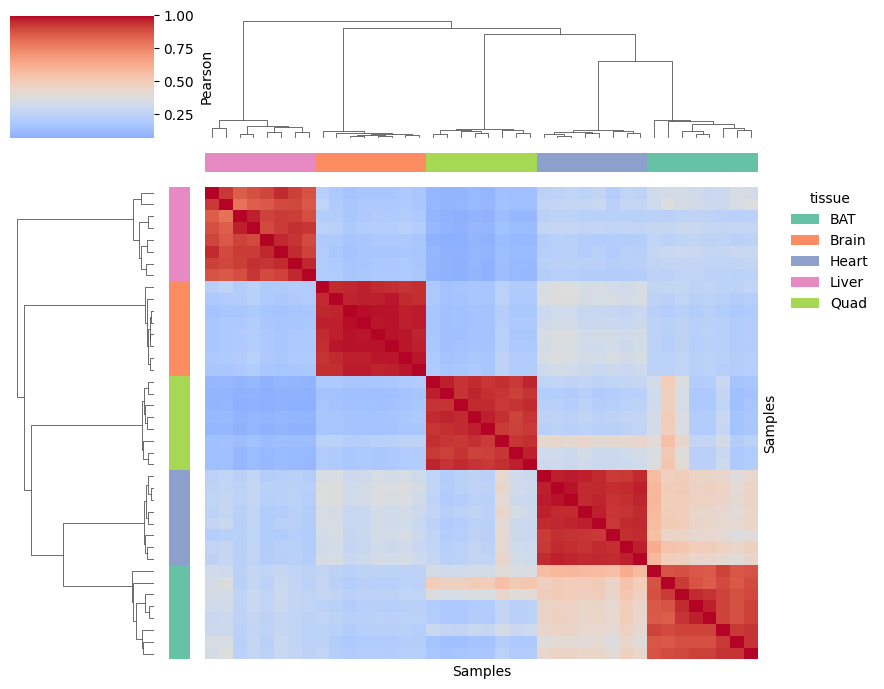
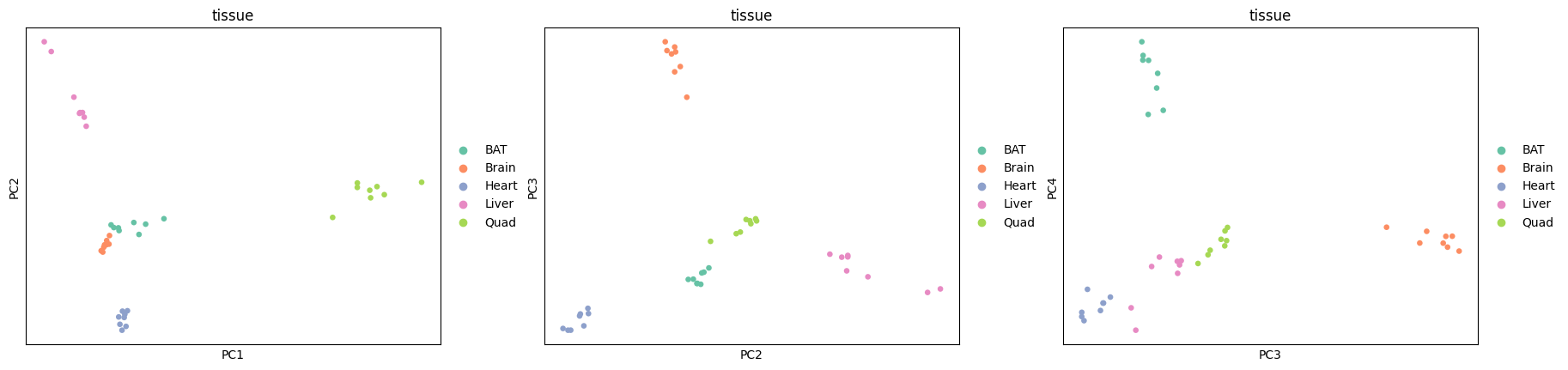
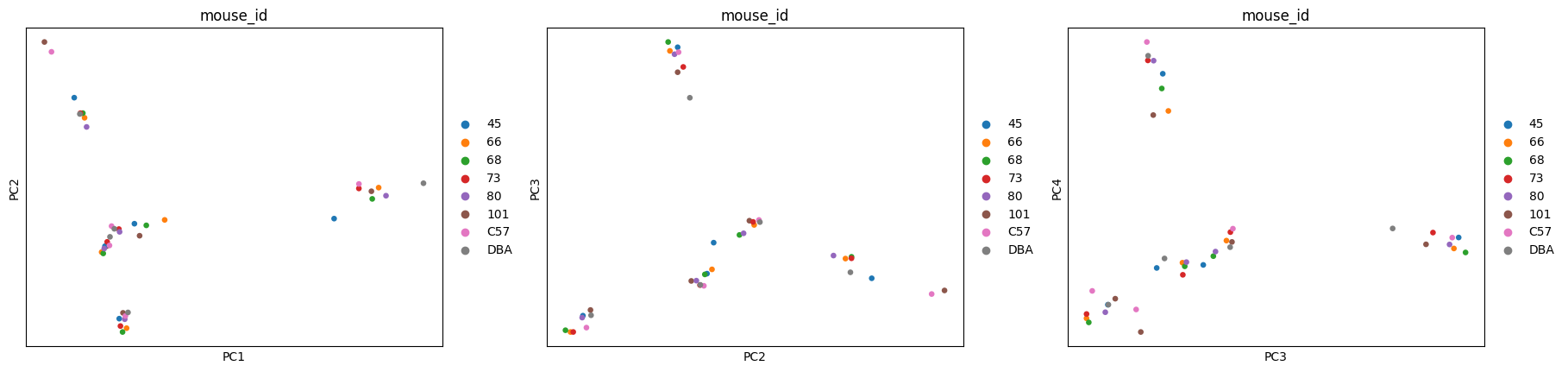
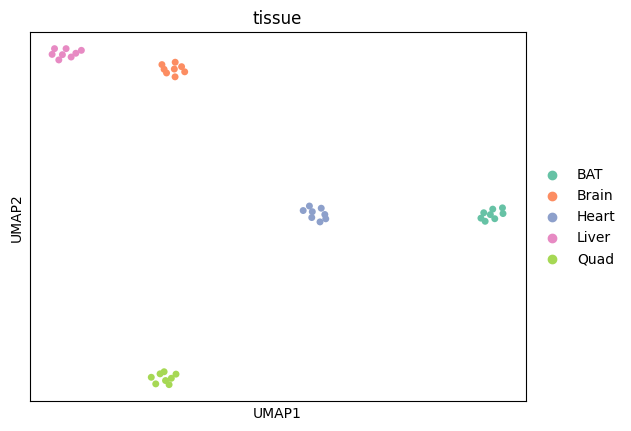
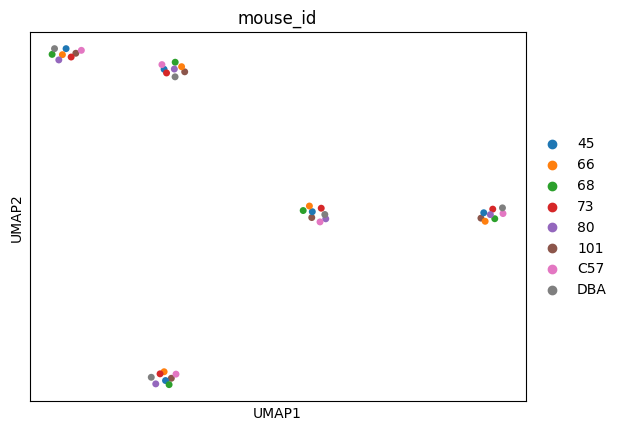
This is further confirmed by both PCA and UMAP, where the tissue is clearly shown as the main source of variation.
PCA
[22]:
sc.tl.pca(adata)
with rc_context({"figure.figsize": (5, 3)}):
sc.pl.pca_variance_ratio(adata, n_pcs=50, log=True)
[23]:
sc.pl.pca(
adata,
color=["tissue", "tissue", "tissue"],
dimensions=[(0, 1), (1, 2), (2, 3)],
ncols=3,
size=90,
palette=adata.uns["colors_tissue"],
)
sc.pl.pca(
adata,
color=["mouse_id", "mouse_id", "mouse_id"],
dimensions=[(0, 1), (1, 2), (2, 3)],
ncols=3,
size=90,
palette=adata.uns["colors_mouse_id"],
)
UMAP
[24]:
sc.pp.neighbors(adata, n_neighbors=4)
sc.tl.umap(adata)
sc.pl.umap(
adata,
color=["tissue"],
size=100,
palette=adata.uns["colors_tissue"],
)
sc.pl.umap(
adata,
color=["mouse_id"],
size=100,
palette=adata.uns["colors_mouse_id"],
)
/home/ifichtner/miniforge3/envs/proteopy-usage2/lib/python3.10/site-packages/sklearn/manifold/_spectral_embedding.py:328: UserWarning: Graph is not fully connected, spectral embedding may not work as expected.
warnings.warn(
Proteoform inference
ProteoPy includes the core functionality of the COPF workflow: (1) compute pairwise peptide correlations between all peptides of a protein, (2) perform hierarchical clustering on the correlation distances, (3) cut each dendrogram into two clusters representing proteoform groups, and (4) score the between- vs. within-cluster correlation difference. Proteins require at least 4 peptides for meaningful clustering.
[25]:
# Require ≥4 peptides per protein for COPF analysis
pr.pp.filter_proteins_by_peptide_count(adata, min_count=4)
pr.pp.remove_zero_variance_vars(adata)
Removed 1770 proteins and 2984 peptides.
Removed 12 variables.
[26]:
# COPF pipeline: correlate, cluster, score
pr.tl.pairwise_peptide_correlations(adata)
pr.tl.peptide_dendograms_by_correlation(adata, method="agglomerative-hierarchical-clustering")
pr.tl.peptide_clusters_from_dendograms(adata, n_clusters=2, min_peptides_per_cluster=2)
pr.tl.proteoform_scores(adata, min_score=0.1, min_pval_adj=0.1)
[27]:
# Remove COPF outlier peptides (cluster_id == 1000000)
copf_outliers = (adata.var["cluster_id"] == 1000000)
adata = adata[:, ~copf_outliers]
Proteoform scores
The pseudo-volcano plot shows proteoform scores vs. adjusted p-values (related to Figure 6B in Bludau et al. (2021)). The two highlighted proteins — Ldb3 (Q9JKS4) and Sorbs2 (Q3UTJ2) — are examined in detail below.
[28]:
bludauI_proteoforms = ["Q9JKS4", "Q3UTJ2"]
pr.pl.proteoform_scores(
adata,
adj=True,
pval_threshold=0.1,
score_threshold=0.1,
log_scores=True,
highlight_prots=bludauI_proteoforms,
)
Looks like you are using a tranform that doesn't support FancyArrowPatch, using ax.annotate instead. The arrows might strike through texts. Increasing shrinkA in arrowprops might help.
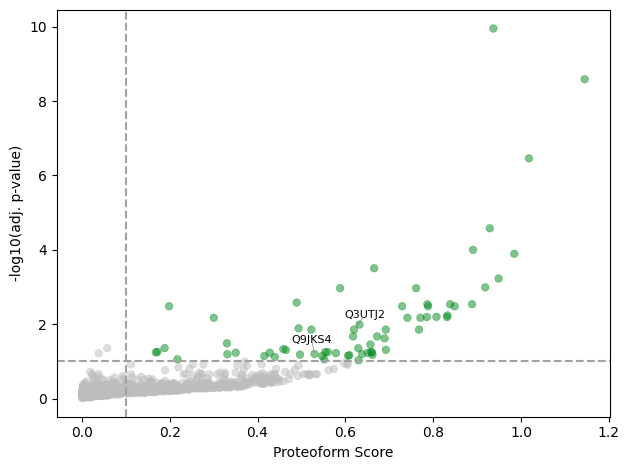
[28]:
<Axes: xlabel='Proteoform Score', ylabel='-log10(adj. p-value)'>
[29]:
pf_df = pr.get.proteoforms_df(adata, only_proteins=True)
n_proteoforms = pf_df[pf_df["is_proteoform"] == 1].shape[0]
print(f"{n_proteoforms} significant proteoforms inferred.")
65 significant proteoforms inferred.
Proteoform exploration
ProteoPy also recovered the two exemplary proteins with proteoform groups highlighted in Bludau et al (2021): LIM domain-binding protein 3 (Ldb3, Q9JKS4) and Sorbin and SH3 domain-containing protein 2 (Sorbs2, Q3UTJ2).
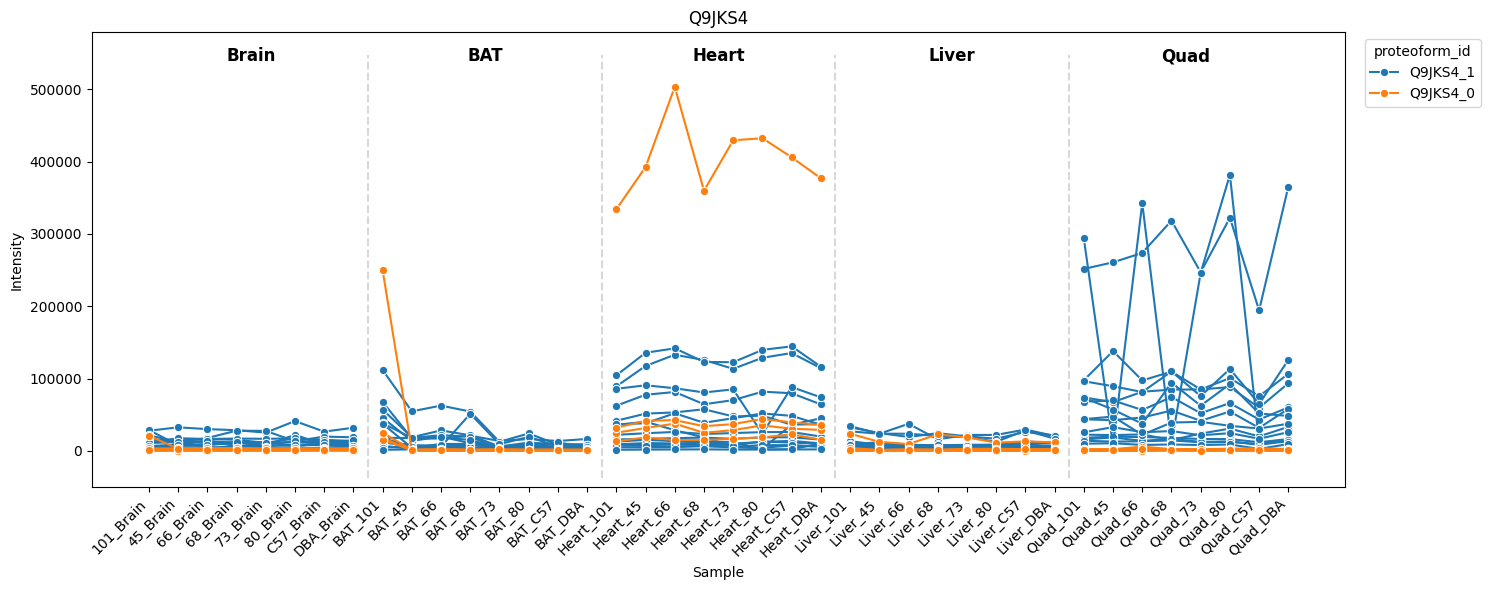
LIM domain-binding protein 3 — Ldb3 (Q9JKS4)
Ldb3 is a muscle-specific protein. COPF assigns its peptides to two tissue-specific proteoform groups, consistent with known alternative splice variants (reproduces Figure 7A).
[30]:
pr.pl.proteoform_intensities(
adata,
protein_ids="Q9JKS4",
order_by="tissue",
order=tissue_order,
xlab_rotation=45,
)
[31]:
pr.get.proteoforms_df(adata, proteins="Q9JKS4")
[31]:
| protein_id | peptide_id | cluster_id | proteoform_score | proteoform_score_pval | proteoform_score_pval_adj | is_proteoform | |
|---|---|---|---|---|---|---|---|
| 0 | Q9JKS4 | QYNNPIGLYSAETLR | 0.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 1 | Q9JKS4 | ASSEGAQGSVSPK | 0.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 2 | Q9JKS4 | VLPGPSQPR | 0.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 3 | Q9JKS4 | EMAQMYQMSLR | 0.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 4 | Q9JKS4 | ILAQMTGTEYMQDPDEEALRR | 1.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 5 | Q9JKS4 | IMGEVMHALR | 1.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 6 | Q9JKS4 | QTWHTTCFVCAACK | 1.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 7 | Q9JKS4 | SKRPIPISTTAPPIQSPLPVIPHQK | 1.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 8 | Q9JKS4 | SASYNLSLTLQK | 1.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 9 | Q9JKS4 | SWHPEEFNCAYCK | 1.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 10 | Q9JKS4 | ASGAGLLGGSLPVKDLAVDSASPVYQAVIK | 1.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 11 | Q9JKS4 | TPLCGHCNNVIR | 1.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 12 | Q9JKS4 | TQSKPEDEADEWARR | 1.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 13 | Q9JKS4 | TSLADVCFVEEQNNVYCER | 1.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 14 | Q9JKS4 | AAQSQLSQGDLVVAIDGVNTDTMTHLEAQNK | 1.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 15 | Q9JKS4 | CYEQFFAPICAK | 1.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 16 | Q9JKS4 | LQGGKDFNMPLTISR | 1.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 17 | Q9JKS4 | GAPAYNPTGPQVTPLAR | 1.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
| 18 | Q9JKS4 | GPFLVAMGR | 1.0 | 0.529813 | 0.002771 | 0.062994 | 1.0 |
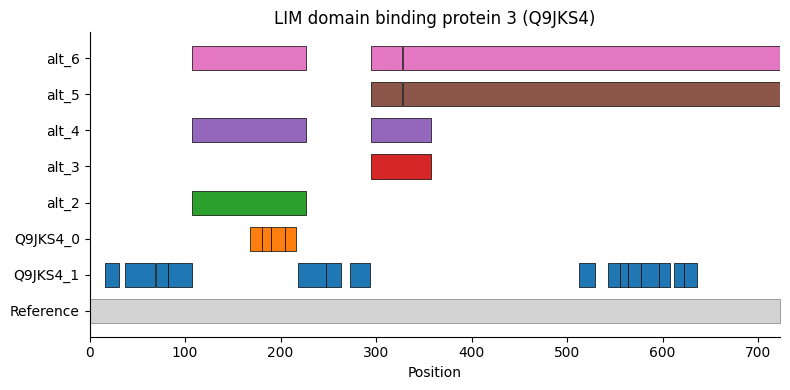
Ldb3 peptide sequence map (reproduces Figure 7C)
Mapping detected peptides onto the canonical protein sequence and annotated UniProt alternative sequences confirms that the two proteoform groups correspond to known splice variants.
[32]:
# Canonical sequence and UniProt alternative sequences for Q9JKS4
# Source: https://www.uniprot.org/uniprotkb/Q9JKS4/entry#sequences
seq = "MSYSVTLTGPGPWGFRLQGGKDFNMPLTISRITPGSKAAQSQLSQGDLVVAIDGVNTDTMTHLEAQNKIKSASYNLSLTLQKSKRPIPISTTAPPIQSPLPVIPHQKDPALDTNGSLATPSPSPEARASPGALEFGDTFSSSFSQTSVCSPLMEASGPVLPLGSPVAKASSEGAQGSVSPKVLPGPSQPRQYNNPIGLYSAETLREMAQMYQMSLRGKASGAGLLGGSLPVKDLAVDSASPVYQAVIKTQSKPEDEADEWARRSSNLQSRSFRILAQMTGTEYMQDPDEEALRRSSTPIEHAPVCTSQATSPLLPASAQSPAAASPIAASPTLATAAATHAAAASAAGPAASPVENPRPQASAYSPAAAASPAPSAHTSYSEGPAAPAPKPRVVTTASIRPSVYQPVPASSYSPSPGANYSPTPYTPSPAPAYTPSPAPTYTPSPAPTYSPSPAPAYTPSPAPNYTPTPSAAYSGGPSESASRPPWVTDDSFSQKFAPGKSTTTVSKQTLPRGAPAYNPTGPQVTPLARGTFQRAERFPASSRTPLCGHCNNVIRGPFLVAMGRSWHPEEFNCAYCKTSLADVCFVEEQNNVYCERCYEQFFAPICAKCNTKIMGEVMHALRQTWHTTCFVCAACKKPFGNSLFHMEDGEPYCEKDYINLFSTKCHGCDFPVEAGDKFIEALGHTWHDTCFICAVCHVNLEGQPFYSKKDKPLCKKHAHAINV"
alt_seqs = {
"Q9JKS4-2_0": {"seq_coord": (107, 227), "group": "alt_2"},
"Q9JKS4-3_0": {"seq_coord": (295, 357), "group": "alt_3"},
"Q9JKS4-4_0": {"seq_coord": (107, 227), "group": "alt_4"},
"Q9JKS4-4_1": {"seq_coord": (295, 357), "group": "alt_4"},
"Q9JKS4-5_0": {"seq_coord": (295, 327), "group": "alt_5"},
"Q9JKS4-5_1": {"seq_coord": (328, 723), "group": "alt_5"},
"Q9JKS4-6_0": {"seq_coord": (107, 227), "group": "alt_6"},
"Q9JKS4-6_1": {"seq_coord": (295, 327), "group": "alt_6"},
"Q9JKS4-6_2": {"seq_coord": (328, 723), "group": "alt_6"},
}
pr.pl.peptides_on_prot_sequence(
adata,
protein_id="Q9JKS4",
group_by="proteoform_id",
ref_sequence=seq,
add_sequences=alt_seqs,
title="LIM domain binding protein 3 (Q9JKS4)",
figsize=(8, 4),
)
[32]:
<Axes: title={'center': 'LIM domain binding protein 3 (Q9JKS4)'}, xlabel='Position'>
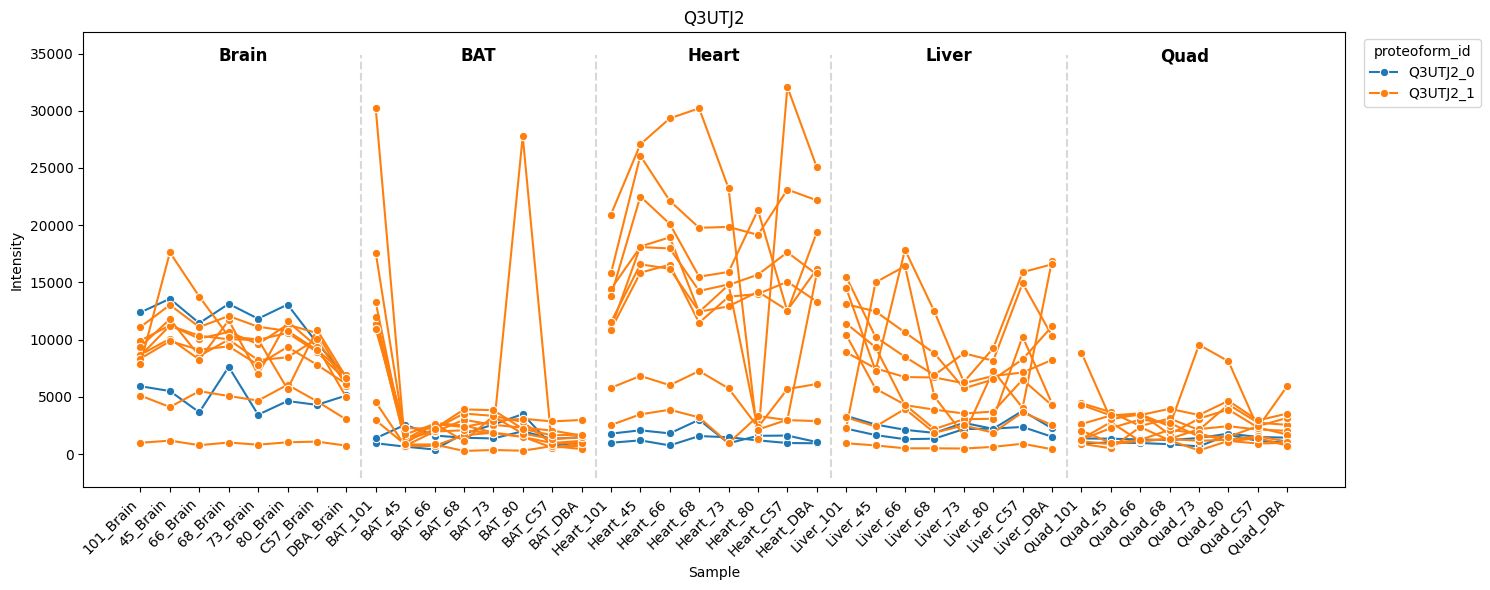
Sorbin and SH3 domain-containing protein 2 — Sorbs2 (Q3UTJ2)
Sorbs2 peptides are assigned to two proteoform groups with distinct tissue expression patterns: one group is abundant across brain, heart, and liver, while the other is brain-specific (reproduces Figure 7D).
[33]:
pr.pl.proteoform_intensities(
adata,
protein_ids="Q3UTJ2",
order_by="tissue",
order=tissue_order,
xlab_rotation=45,
)
[34]:
pr.get.proteoforms_df(adata, proteins="Q3UTJ2")
[34]:
| protein_id | peptide_id | cluster_id | proteoform_score | proteoform_score_pval | proteoform_score_pval_adj | is_proteoform | |
|---|---|---|---|---|---|---|---|
| 0 | Q3UTJ2 | LAFLVSPVPFR | 0.0 | 0.619637 | 0.000343 | 0.01393 | 1.0 |
| 1 | Q3UTJ2 | ASVVEALDSALKDICDQIK | 0.0 | 0.619637 | 0.000343 | 0.01393 | 1.0 |
| 2 | Q3UTJ2 | QGIFPVSYVEVVKR | 1.0 | 0.619637 | 0.000343 | 0.01393 | 1.0 |
| 3 | Q3UTJ2 | APHYPGIGPVDESGIPTAIR | 1.0 | 0.619637 | 0.000343 | 0.01393 | 1.0 |
| 4 | Q3UTJ2 | SFISSSPSSPSR | 1.0 | 0.619637 | 0.000343 | 0.01393 | 1.0 |
| 5 | Q3UTJ2 | SIFEYEPGK | 1.0 | 0.619637 | 0.000343 | 0.01393 | 1.0 |
| 6 | Q3UTJ2 | AQPARPPPPVQPGEIGEAIAK | 1.0 | 0.619637 | 0.000343 | 0.01393 | 1.0 |
| 7 | Q3UTJ2 | SYSSTLTDLGR | 1.0 | 0.619637 | 0.000343 | 0.01393 | 1.0 |
| 8 | Q3UTJ2 | VGIFPISYVEK | 1.0 | 0.619637 | 0.000343 | 0.01393 | 1.0 |
| 9 | Q3UTJ2 | ADLPGSSSTFTK | 1.0 | 0.619637 | 0.000343 | 0.01393 | 1.0 |
| 10 | Q3UTJ2 | GLGDQSSSR | 1.0 | 0.619637 | 0.000343 | 0.01393 | 1.0 |
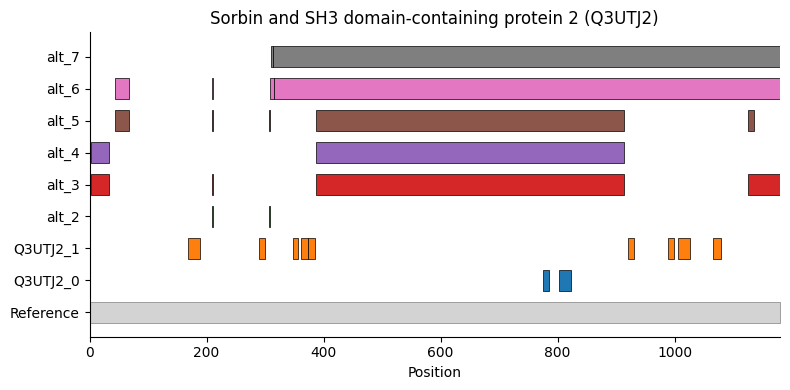
Sorbs2 peptide sequence map (reproduces Figure 7F)
The brain-specific proteoform group maps to a region covered by alternative sequences 3, 4 and 5 in UniProt (10 in Bludau et. al. Figure 7F), a brain-specific splice variant.
[35]:
# Canonical sequence and UniProt alternative sequences for Q3UTJ2
# Source: https://www.uniprot.org/uniprotkb/Q3UTJ2/entry#sequences
seq = "MNTDSGGCARKRAAMSVTLTSVKRVQSSPNLLAAGRESQSPDSAWRSYNDRNPETLNGDATYSSLAAKGFRSVRPNLQDKRSPTQSQITINGNSGGAVSPVSYYQRPFSPSAYSLPASLNSSIIMQHGRSLDSAETYSQHAQSLDGTMGSSIPLYRSSEEEKRVTVIKAPHYPGIGPVDESGIPTAIRTTVDRPKDWYKTMFKQIHMVHKPGLYNSPYSAQSHPAAKTQTYRPLSKSHSDNGTDAFKEVPSPVPPPHVPPRPRDQSSTLKHDWDPPDRKVDTRKFRSEPRSIFEYEPGKSSILQHERPVSIYQSSIDRSLERPSSSASMAGDFRKRRKSEPAVGPLRGLGDQSSSRTSPGRADLPGSSSTFTKSFISSSPSSPSRAQGGDDSKMCPPLCSYSGLNGTPSGELECCNAYRQHLDVPGDSQRAITFKNGWQMARQNAEIWSSTEETVSPKIKSRSCDDLLNDDCDSFPDPKTKSESMGSLLCEEDSKESCPMTWASPYIQEVCGNSRSRLKHRSAHNAPGFLKMYKKMHRINRKDLMNSEVICSVKSRILQYEKEQQHRGLLHGWSQSSTEEVPRDVVPTRISEFEKLIQKSKSMPNLGDEMLSPITLEPPQNGLCPKRRFSIESLLEEETQVRHPSQGQRSCKSNTLVPIHIEVTSDEQPRTHMEFSDSDQDGVVSDHSDYVHVEGSSFCSESDFDHFSFTSSESFYGSSHHHHHHHHHHRHLISSCKGRCPASYTRFTTMLKHERAKHENMDRPRRQEMDPGLSKLAFLVSPVPFRRKKILTPQKQTEKAKCKASVVEALDSALKDICDQIKAEKRRGSLPDNSILHRLISELLPQIPERNSSLHALKRSPMHQPFHPLPPDGASHCPLYQNDCGRMPHSASFPDVDTTSNYHAQDYGSALSLQDHESPRSYSSTLTDLGRSASRERRGTPEKEKLPAKAVYDFKAQTSKELSFKKGDTVYILRKIDQNWYEGEHHGRVGIFPISYVEKLTPPEKAQPARPPPPVQPGEIGEAIAKYNFNADTNVELSLRKGDRIILLKRVDQNWYEGKIPGTNRQGIFPVSYVEVVKRNAKGAEDYPDPPLPHSYSSDRIYTLSSNKPQRPGFSHENIQGGGEPFQALYNYTPRNEDELELRESDVVDVMEKCDDGWFVGTSRRTKFFGTFPGNYVKRL"
alt_seqs = {
"Q3UTJ2-2_0": {"seq_coord": (210, 211), "group": "alt_2"},
"Q3UTJ2-2_1": {"seq_coord": (307, 308), "group": "alt_2"},
"Q3UTJ2-3_0": {"seq_coord": (3, 34), "group": "alt_3"},
"Q3UTJ2-3_1": {"seq_coord": (210, 211), "group": "alt_3"},
"Q3UTJ2-3_2": {"seq_coord": (387, 914), "group": "alt_3"},
"Q3UTJ2-3_3": {"seq_coord": (1125, 1180), "group": "alt_3"},
"Q3UTJ2-4_0": {"seq_coord": (3, 34), "group": "alt_4"},
"Q3UTJ2-4_1": {"seq_coord": (387, 914), "group": "alt_4"},
"Q3UTJ2-5_0": {"seq_coord": (44, 67), "group": "alt_5"},
"Q3UTJ2-5_1": {"seq_coord": (210, 211), "group": "alt_5"},
"Q3UTJ2-5_2": {"seq_coord": (307, 308), "group": "alt_5"},
"Q3UTJ2-5_3": {"seq_coord": (387, 914), "group": "alt_5"},
"Q3UTJ2-5_4": {"seq_coord": (1125, 1135), "group": "alt_5"},
"Q3UTJ2-6_0": {"seq_coord": (44, 67), "group": "alt_6"},
"Q3UTJ2-6_1": {"seq_coord": (210, 211), "group": "alt_6"},
"Q3UTJ2-6_2": {"seq_coord": (308, 316), "group": "alt_6"},
"Q3UTJ2-6_3": {"seq_coord": (316, 1180), "group": "alt_6"},
"Q3UTJ2-7_0": {"seq_coord": (310, 313), "group": "alt_7"},
"Q3UTJ2-7_1": {"seq_coord": (313, 1180), "group": "alt_7"},
}
pr.pl.peptides_on_prot_sequence(
adata,
protein_id="Q3UTJ2",
group_by="proteoform_id",
ref_sequence=seq,
add_sequences=alt_seqs,
title="Sorbin and SH3 domain-containing protein 2 (Q3UTJ2)",
figsize=(8, 4),
)
[35]:
<Axes: title={'center': 'Sorbin and SH3 domain-containing protein 2 (Q3UTJ2)'}, xlabel='Position'>
Proteoform quantification and statistical analysis
To test if the inferred proteoform groups are indeed tissue-specific, a one-way ANOVA analysis can be performed directly using ProteoPy functions. In strong agreement with Bludau et al. (2021), our analysis revealed that 59 out of 65 proteoform-containing proteins (90.8%) are expressed in a tissue-specific manner.
[36]:
# Aggregate peptide intensities to proteoform level
adata_pfs = adata.copy()
pr.pp.quantify_proteoforms(adata_pfs, group_by="proteoform_id")
[37]:
# Retain only significant proteoforms (score >= 0.1, adj. p-value <= 0.1)
pf_mask = (
(adata_pfs.var["proteoform_score"].astype(float) >= 0.1)
& (adata_pfs.var["proteoform_score_pval_adj"].astype(float) <= 0.1)
)
adata_pfs = adata_pfs[:, pf_mask].copy()
[38]:
# Log2 transform for statistical testing
adata_pfs.layers["raw"] = adata_pfs.X
adata_pfs.X[adata_pfs.X == 0] = np.nan
adata_pfs.X = np.log2(adata_pfs.X)
adata_pfs.X[np.isnan(adata_pfs.X)] = 0
[39]:
# One-way ANOVA across tissues
pr.tl.differential_abundance(
adata_pfs,
method="anova_oneway",
multitest_correction="bonferroni",
group_by="tissue",
alpha=0.01,
space="log",
)
Saved test results in .varm['anova_oneway;tissue;all']
[40]:
anova_results = pr.get.differential_abundance_df(adata_pfs, keys="anova_oneway;tissue;all")
anova_results.rename(columns={"var_id": "proteoform_id"}, inplace=True)
anova_results
[40]:
| proteoform_id | test_type | group_by | design | fstat | pval | mean_Brain | mean_BAT | mean_Heart | mean_Liver | mean_Quad | pval_adj | is_diff_abundant | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | O08601_0 | anova_oneway | tissue | all | 29.998692 | 7.135979e-11 | 15.447875 | 16.438822 | 13.642355 | 15.286296 | 13.226680 | 9.276772e-09 | True |
| 1 | O08601_1 | anova_oneway | tissue | all | 309.316738 | 8.846917e-27 | 17.173194 | 16.274997 | 16.297549 | 19.401295 | 16.539176 | 1.150099e-24 | True |
| 2 | O54724_0 | anova_oneway | tissue | all | 12.603239 | 1.878084e-06 | 15.879478 | 13.639894 | 8.489357 | 10.545068 | 11.828292 | 2.441509e-04 | True |
| 3 | O54724_1 | anova_oneway | tissue | all | 184.250825 | 5.444913e-23 | 16.359031 | 19.356209 | 18.207868 | 15.845380 | 17.226098 | 7.078387e-21 | True |
| 4 | O54749_0 | anova_oneway | tissue | all | 5.288221 | 1.938883e-03 | 13.162980 | 12.879554 | 14.428646 | 13.568651 | 13.071253 | 2.520548e-01 | False |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 125 | Q9JKS4_1 | anova_oneway | tissue | all | 66.936903 | 6.594055e-16 | 17.255804 | 17.226273 | 19.196674 | 17.040296 | 19.944539 | 8.572271e-14 | True |
| 126 | Q9QYR6_0 | anova_oneway | tissue | all | 14.856602 | 3.441306e-07 | 15.506743 | 12.560879 | 13.676294 | 13.230452 | 13.061866 | 4.473698e-05 | True |
| 127 | Q9QYR6_1 | anova_oneway | tissue | all | 646.764070 | 2.861598e-32 | 20.504493 | 16.481401 | 16.861090 | 17.344528 | 16.848407 | 3.720078e-30 | True |
| 128 | Q9Z204_0 | anova_oneway | tissue | all | 75.275766 | 1.071660e-16 | 16.117296 | 12.403909 | 12.737527 | 13.374945 | 12.716591 | 1.393159e-14 | True |
| 129 | Q9Z204_1 | anova_oneway | tissue | all | 152.715690 | 1.224770e-21 | 15.790335 | 14.611674 | 13.502690 | 16.977432 | 13.218752 | 1.592201e-19 | True |
130 rows × 13 columns
[41]:
# Count proteins with all proteoforms being significantly tissue-specific
protein_id_map = adata_pfs.var[["protein_id", "protein_id_old"]].set_index("protein_id")["protein_id_old"]
anova_results["protein_id"] = anova_results["proteoform_id"].map(protein_id_map)
n_tissue_specific_pfs = anova_results.groupby("protein_id")["is_diff_abundant"].all().sum()
print(f"{n_tissue_specific_pfs} tissue-specific proteoform groups found via ANOVA.")
59 tissue-specific proteoform groups found via ANOVA.
Summary
This notebook reproduced the core proteoform inference workflow from Bludau et al. (2021) using ProteoPy. Key results:
Proteoform detection: COPF identified 65 proteins with proteoform groups, comparable to the 63 reported in the original study (Figure 6B).
Biological verification: The tissue-specific proteoform assignments for Ldb3 (Q9JKS4) and Sorbs2 (Q3UTJ2) match the published findings (Figures 7A, 7C, 7D, 7F), with peptide-to-sequence mappings consistent with known alternative splice variants.
Tissue specificity: ANOVA analysis recovered 59 tissue-specific proteoform groups (90.8% of the inferred proteoform groups) — similar to the 56 tissue-specific proteoform groups (88.9% of the inferred proteoform groups) reported by Bludau et al.
Minor discrepancies between reported numbers are expected due to implementation details in preprocessing (e.g., peptide modification summarization, overlapping peptide handling) and statistical testing.
[42]:
!pip freeze
adjustText==1.3.0
anndata==0.11.4
anyio==4.12.1
argon2-cffi==25.1.0
argon2-cffi-bindings==25.1.0
array-api-compat==1.13.0
arrow==1.4.0
asttokens==3.0.1
async-lru==2.2.0
attrs==25.4.0
babel==2.18.0
beautifulsoup4==4.14.3
biopython==1.86
bleach==6.3.0
certifi==2026.1.4
cffi==2.0.0
charset-normalizer==3.4.4
comm==0.2.3
contourpy==1.3.2
cycler==0.12.1
debugpy==1.8.20
decorator==5.2.1
defusedxml==0.7.1
et_xmlfile==2.0.0
exceptiongroup==1.3.1
executing==2.2.1
fastjsonschema==2.21.2
fonttools==4.61.1
fqdn==1.5.1
h11==0.16.0
h5py==3.15.1
httpcore==1.0.9
httpx==0.28.1
idna==3.11
igraph==1.0.0
ipykernel==7.2.0
ipython==8.38.0
isoduration==20.11.0
jedi==0.19.2
Jinja2==3.1.6
joblib==1.5.3
json5==0.13.0
jsonpointer==3.0.0
jsonschema==4.26.0
jsonschema-specifications==2025.9.1
jupyter-events==0.12.0
jupyter-lsp==2.3.0
jupyter_client==8.8.0
jupyter_core==5.9.1
jupyter_server==2.17.0
jupyter_server_terminals==0.5.4
jupyterlab==4.5.4
jupyterlab_pygments==0.3.0
jupyterlab_server==2.28.0
kiwisolver==1.4.9
lark==1.3.1
legacy-api-wrap==1.5
llvmlite==0.46.0
MarkupSafe==3.0.3
matplotlib==3.10.8
matplotlib-inline==0.2.1
mistune==3.2.0
natsort==8.4.0
nbclient==0.10.4
nbconvert==7.17.0
nbformat==5.10.4
nest-asyncio==1.6.0
networkx==3.4.2
notebook_shim==0.2.4
numba==0.64.0
numpy==2.2.6
openpyxl==3.1.5
overrides==7.7.0
packaging @ file:///home/conda/feedstock_root/build_artifacts/bld/rattler-build_packaging_1769093650/work
pandas==2.3.3
pandocfilters==1.5.1
parso==0.8.6
patsy==1.0.2
pexpect==4.9.0
pillow==12.1.1
platformdirs==4.9.2
pooch==1.9.0
prometheus_client==0.24.1
prompt_toolkit==3.0.52
-e git+https://github.com/UKHD-NP/proteopy.git@05b165881b1465933b5ffe86e3acbb30abc2b6c0#egg=proteopy
psutil==7.2.2
ptyprocess==0.7.0
pure_eval==0.2.3
pycparser==3.0
Pygments==2.19.2
pynndescent==0.6.0
pyparsing==3.3.2
python-dateutil==2.9.0.post0
python-igraph==1.0.0
python-json-logger==4.0.0
pytz==2025.2
PyYAML==6.0.3
pyzmq==27.1.0
referencing==0.37.0
requests==2.32.5
rfc3339-validator==0.1.4
rfc3986-validator==0.1.1
rfc3987-syntax==1.1.0
rpds-py==0.30.0
scanpy==1.11.5
scikit-learn==1.7.2
scipy==1.15.3
seaborn==0.13.2
Send2Trash==2.1.0
session-info2==0.4
six==1.17.0
soupsieve==2.8.3
stack-data==0.6.3
statsmodels==0.14.6
terminado==0.18.1
texttable==1.7.0
threadpoolctl==3.6.0
tinycss2==1.4.0
tomli==2.4.0
tornado==6.5.4
tqdm==4.67.3
traitlets==5.14.3
typing_extensions==4.15.0
tzdata==2025.3
umap-learn==0.5.11
uri-template==1.3.0
urllib3==2.6.3
wcwidth==0.6.0
webcolors==25.10.0
webencodings==0.5.1
websocket-client==1.9.0